

IQIRVO ELATIVE trial
Efficacy

Adapted from Kowdley et al. 2024.
Biochemical response was analysed across patient subgroups3
The biochemical response of IQIRVO + UDCA was observed across subgroups irrespective of age, pruritus symptoms, ALP level and advanced disease stage*†3
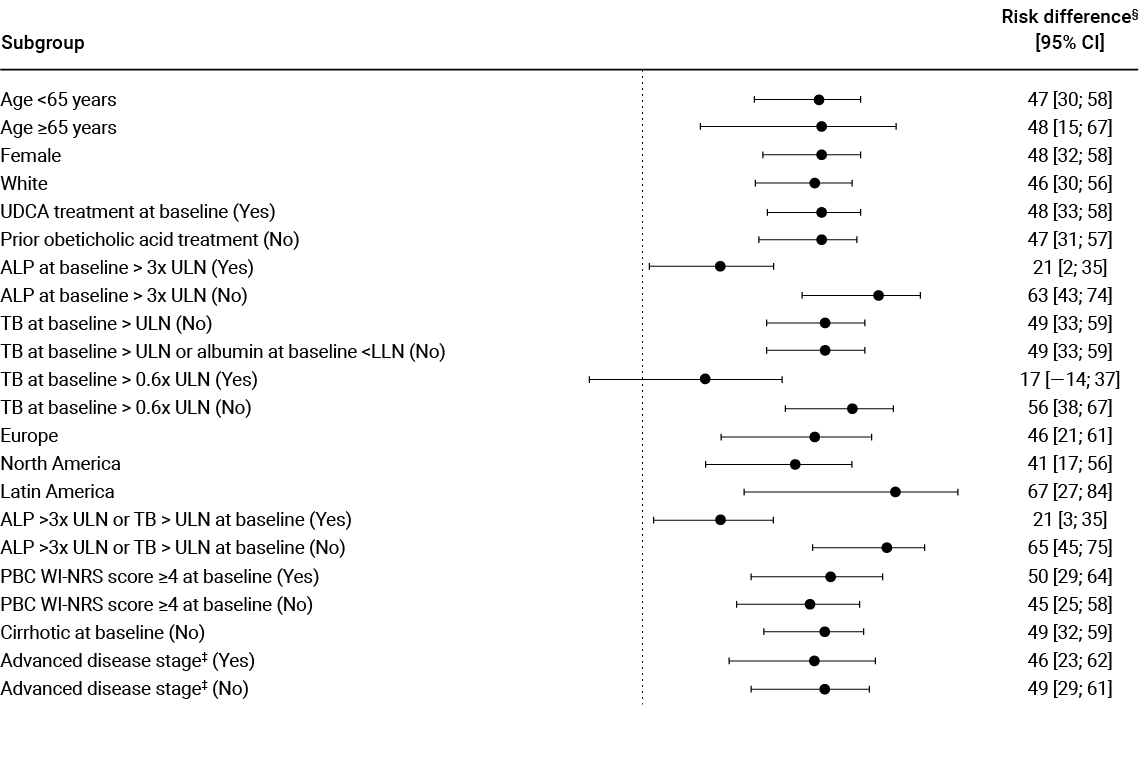
Adapted from Kowdley et al. 2024.
SECONDARY ENDPOINT:
ALP reduction¶1,2
ALP reduction from baseline in just 4 weeks –sustained through to 52 weeks†1,2
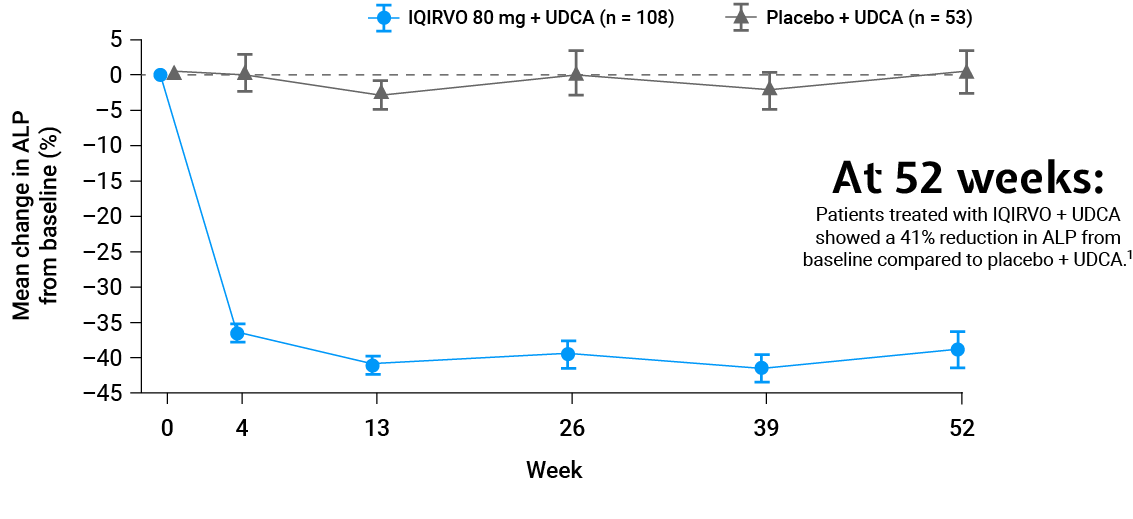
Adapted from Kowdley et al. 2024.
SECONDARY ENDPOINT:
ALP normalisation¶1,2
ALP normalisation was achieved with 15% of patients treated with IQIRVO + UDCA vs Placebo + UDCA†1,2
Mean reduction to achieve ALP normalisation with IQIRVO + UDCA was 218 U/L1,2

Adapted from Kowdley et al. 2024.
Mean baseline ALP: 322 U/L1,2
ULN definition: 104 U/L for women 154/161 (96%)# participants1
IQIRVO: Impact of pruritus on patients’ quality of life¶1,2
IQRIVO + UDCA demonstrated an observed mean change in pruritus from baseline through Week 52 and Week 24 as measured by PBC WI-NRS in those with moderate-to-severe pruritus at baseline when compared to placebo + UDCA (this result was non-significant)†**1,2
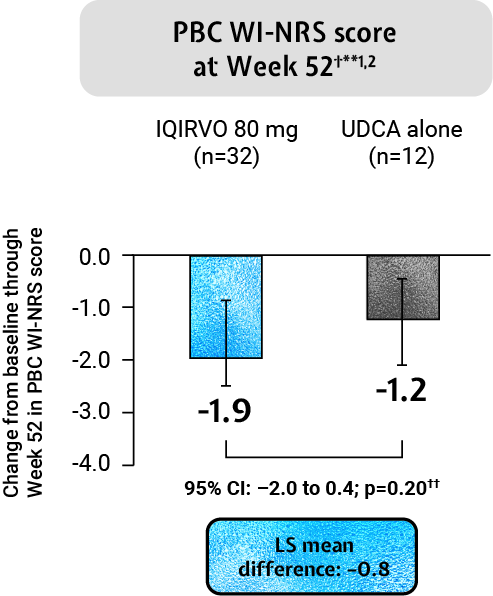
Adapted from Kowdley et al. 2024.
IQIRVO + UDCA reduced the impact of pruritus on QoL vs placebo + UDCA as measured by the PBC-40 itch domain and 5-D itch scale†**1,2
The PBC-40 is a patient-derived, disease specific QoL measure developed and validated for use in PBC4
The 5-D Itch scale is a measure of itching that has been validated in patients with chronic pruritus5
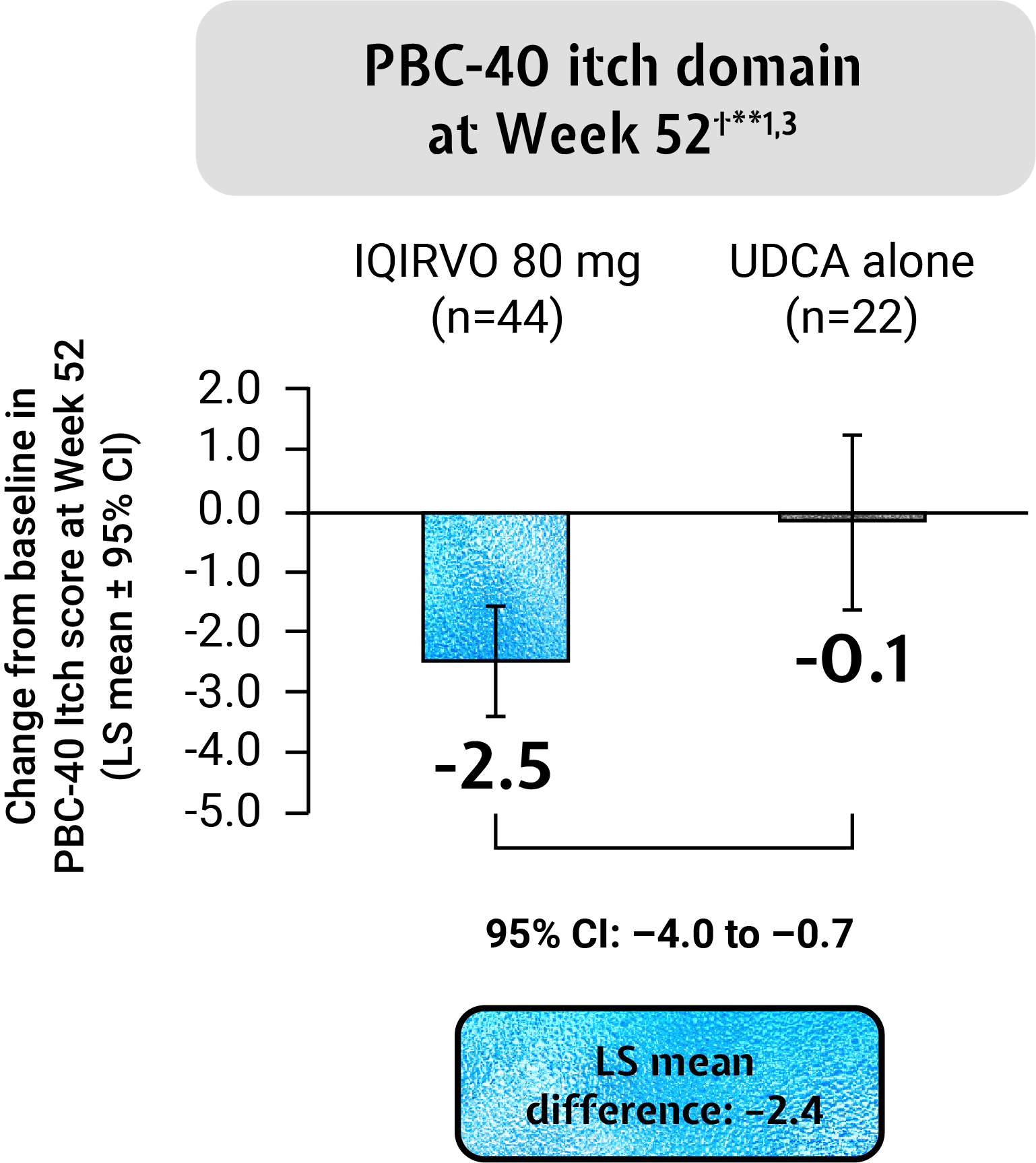

Adapted from Kowdley et al. 2024.
BASELINE ALP SUBGROUP ANALYSIS
Biochemical response was achieved in 71% of patients with ALP ≤3 x ULN and 21% of patients with ALP >3x ULN at baseline*6
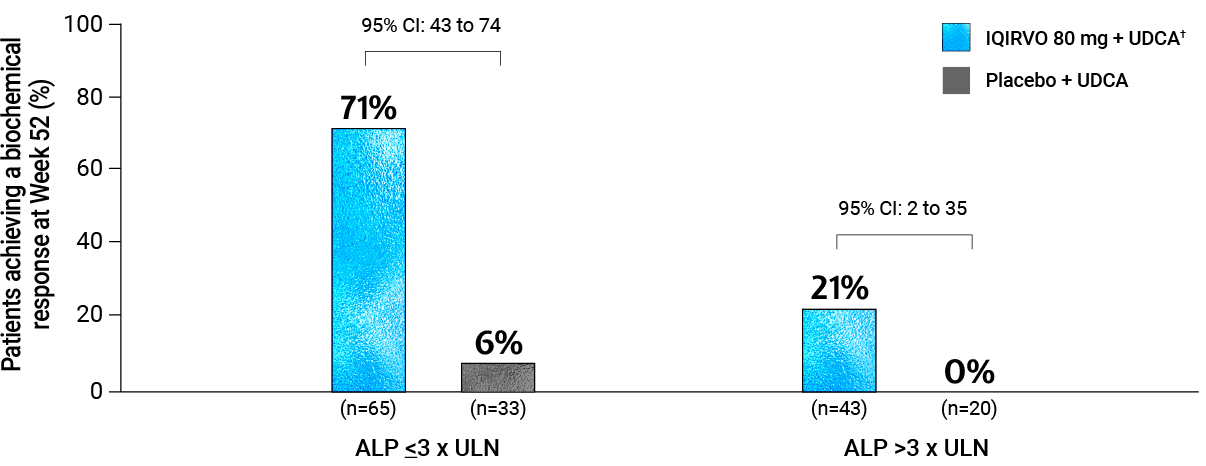
Adapted from Ipsen Data on File
Footnotes
*Biochemical response is defined in the trial as ALP <1.67 x ULN, and ALP decrease ≥15% and TB ≤ULN at 52 weeks.1
†Patients either received IQIRVO on a background of UDCA (102/108, 94%) or received UDCA plus a placebo (51/53, 96%).1
‡Defined as liver stiffness at baseline >10.0 kPa and/or bridging fibrosis or cirrhosis on histology.3
§Risk difference corresponds to difference (%) in response (IQIRVO + UDCA vs placebo + UDCA). If the subgroup at baseline included fewer than 20 patients across treatment groups or fewer than 5 patients for a treatment group, the subgroup was omitted.3
¶Key secondary endpoints (normalisation of ALP at Week 52, and the change from baseline in the WI-NRS score through Week 52 and through Week 24) were assessed with the use of a pre-specified fixed sequence testing approach, at a two-sided alpha of 0.05, until a non-significant result was encountered. Other secondary endpoints are reported as point estimates and 95% confidence intervals, which were not adjusted for multiple testing.1
#Percent of the study population.1
**In patients with moderate-to-severe pruritus (defined as PBC WI-NRS Score ≥4).1
Abbreviations
ALP, alkaline phosphatase; CI, confidence interval; kPa, kilopascals; LLN, lower limit of normal; mg, milligram; PBC WI-NRS, primary biliary cholangitis worst itch numeric rating scale; QoL, quality of life; TB, total bilirubin; UDCA, ursodeoxycholic acid; ULN, upper limit of normal; U/L, units per litre.
References
- Kowdley KV et al. N Engl J Med. 2024;390(9):795–805.
- IQIRVO® (elafibranor) Summary of product characteristics (SmPC). 2024.
- Kowdley KV et al. Supplement to: N Engl J Med. 2024;390(9):795–805.
- Jacoby A et al. Gut. 2005;54(11):1622–1629.
- Elmen S et al. Br J Dermatol 2010;162:587–59.
- Ipsen Data on File ELA-ALL-000985.
Resources
- PBC
- IQIRVO MoA
- The ELATIVE trial: Study design & baseline characteristics
- The ELATIVE trial: Efficacy
- The ELATIVE trial: Safety data
- IQIRVO Safety Data
- Dosing
- Resources
Contact your Ipsen representative
To access relevant information about Primary Biliary Cholangitis, please confirm the following:
▼This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. Reporting of side effects. If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in the package leaflet. You can also report side effects directly via the Yellow Card Scheme at https://yellowcard.mhra.gov.uk/ or search for MHRA Yellow Card in Google Play or Apple App Store. Side effects should also be reported to Ipsen via email at pharmacovigilance.uk-ie@ipsen.com or phone 01753 627777. By reporting side effects, you can help provide more information on the safety of this medicine.